
Formatting Clinical Evaluation Reports
A Whitepaper by Charles Hurwitz and Nancy J Stark.
©2015 Clinical Device Group Inc, all rights reserved.
Add us to your Safe Senders list to assure continued receipt.
Want to stay on the mailing list? Opt-in for
whitepapers.
A number of clients have commented to me this year that Notified Bodies are rejecting their Clinical Evaluation Reports for nonconformance to the formatting rules of MEDDEV 2.7.1 r3. Some companies are undertaking complete rewrites of their existing clinical evaluations, so concerned are they that the reports will be rejected at the next review cycle. The EU intends to change the formatting rules, however my European colleagues inform me the 2009 version of the guideline is still firmly in effect and that changes are likely to come slowly. In this whitepaper we'll review the format suggested by MEDDEV 2.7.1 r3 and look at some common formatting errors. [1. MEDDEV 2.7.1 rev 3]
$$$ Clinical Device Group, A Medical Device Consultancy $$$ Clinical Evaluations
|
A Muddled History
It was people at the Cochrane Library, the US Agency for Healthcare Research and Quality (AHRQ), and the National Library of Congress who first worked with the problem of how to use the existing literature to evaluate safety, efficacy, or performance of products and procedures. The Global Harmonization Task Force Study Group 5 (now defunct) published a document titled "Clinical Evaluation" in May of 2007. (You may download a copy for free from CDG's Vintage Guidances web page.) The Active Implantibles Medical Device Directive of 1990 discussed the essential requirement of clinical evaluations in Annex 7. The Medical Device Directive of 1993 discussed the essential requirement of clinical evaluations in Annex X. But neither directive gave much information about format. Both directives were updated in March 2010 with Directive 2007/47/EC, strengthening the requirement for clinical evaluations and their content, but without much regard to format, saying only that the clinical evaluation shall be documented as a report, regularly updated, and signed appropriately.
[2. Directive 2007/47/EC]
Suggested Format from MEDDEV 2.7.1 r3 (2009)
In the year or two following the 2010 revision of the Medical Device Directive many of the Notified Bodies (NB) themselves didn't know how a clinical evaluation report should be written or what their authority was in terms of reviewing them. Any CER that bore even a vague resemblance to what was described in the Directive was deemed to be acceptable. The variety of formats that have crossed our desks has been remarkable.
However as time has passed the NBs have better understood their role in the evaluation process. They have formed their own professional society, the Notified Body Operations Group (NBOG), and have issued best practice guides for their members. With the issuance of a checklist for an audit of a Notified Body's review process, NBs are being held accountable for their decisions and are refusing to accept reports that do not correspond strictly to the MEDDEV 2.7.1 r3 guideline. [3. NBOG CL 2010-1]
Appendix E of the MEDDEV guideline is "a possible" format for a clinical evaluation report. This is the format you are expected to follow for compliance. Reviewers work with a checklist at the back of the guideline when reviewing a CER and if a section or item is missing you may get a nonconformity, costing extra weeks of review time and lost sales opportunities. What follows is a review of the recommended format for a CER. [4. MEDDEV 2.7.1 r3 p32]
1. General Details—A Three-Legged Stool
Think of the clinical evaluation report as a three-legged stool. One leg is the literature review, a second leg is the assessment of your risk management process, and the third leg is the assessment of any clinical investigations your firm has sponsored.
The three "legs" are brought together as attachments to a finalized report, the "seat" of the stool. In the opening paragraphs of the seat you must take care to identify exactly what device(s) are covered by the report; identifying the devices by proprietary name, any code names assigned during device development (one company used the code name "Kermit" because they intended to leap over the competition,) and the manufacturer(s) of the device.
Your firm should assign (or outsource) a medical writer who will act as project manager for preparing the report, have authority to obtain and inspect all necessary records and reports, and have authority to work with external services as needed. In our experience as consultants, it takes about 140 hours of labor over a three month duration to write a clinical evaluation.
2. Description of the device and its intended application in or on the body
Provide a concise physical description of the device, cross referencing to relevant sections of the manufacturer’s technical information as appropriate. The description should cover information such as:
• materials, including whether the device incorporates a medicinal substance (already on the market or new), tissues, or blood products;
• the device components, including software and accessories;
• mechanical characteristics; and
• other characteristics, such as sterile vs. non-sterile, radioactive, and the like.
You should think like the sales department of a successful department store: "there shall be no barriers to customer buying." In a CER case, there should be no barriers to the Notified Body quickly understanding your technology. Anticipate the questions a naive reviewer will have and address the questions in the seat of the report. It's useful to include diagrams, drawings, photographs, comparative tables, lists of animal safety testing, text boxes, and the like. These tools help the reviewer to grasp the essense of your technology without doing a lot of independent research.
Don't forget to state the intended application of the device – single use or reusable; invasive or non invasive; implantable or not; duration of use or contact with the body; organs, tissues or body fluids contacted by the device; is the device used in the home – and describe how the device achieves its intended purposes.
Sometimes clients forget about the processing aids or biological substances used during the manufacturing process. Metal parts are routinely washed with solvents to remove oily residues, biological products may have their origin from unidentified sources of tissue (fish from the sea.) Sometimes the final processing takes place in the physician's office or lab. You should be able to demonstrate the absence of any inadvertent residue, or that any such residue does not interfere with safety or performance.
3. Intended therapeutic and/or diagnostic indications and claims
Devices are physically applied on or in the body for a reason: to treat, mitigate, cure or diagnose. You must clearly state the medical conditions (indications) and discuss the medical background for each condition. Reviewers are not experts in all fields of medicine. Your goal as the medical writer is to remove all barriers to clarity, understanding, and full information.
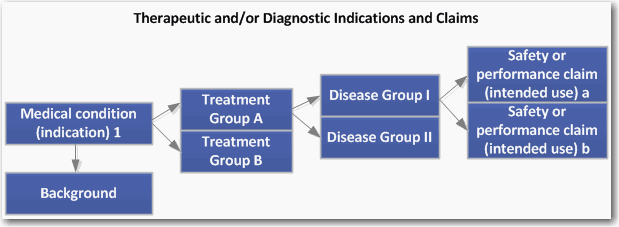
You then identify the target treatment groups and diseases for the medical conditions. Who are the patient groups that will be exposed to the device? Let's say the medical condition is pregnancy. The likely patient group is young, healthy women with no concomitant illnesses or genetic variants.
What are the diseases or disease subgroups for the medical conditions? Again, suppose the medical condition is pregnancy. You may be targeting pregnant women with diabetes, preeclampsia, or other issues. Finally, list the specific safety or performance claims (intended uses) you will make for the device and defend them.
You will have some strategic issues to deal with along the way. For example, do you want to mix device indications and, say, cosmetic indications in the same clinical evaluation? Or is it a smarter to submit an evaluation for medical use today and amend it for cosmetic use in the future?
4. Context of the evaluation
Outline the developmental context for the device. The information should include whether the device is based on a new technology, a new clinical application of an existing technology, or the result of incremental change of an existing technology. The amount of information will differ according to the history of the technology. Where a completely new technology has been developed, this section would need to give an overview of the developmental process and the points in the development cycle at which clinical data have been generated. For example, a manufacturer developed an resorbable container in which a patient's tissue was held in place until it could take hold and grow, as opposed to being held in place with a sling of sutures. The CER included data about both the resorbable container and preparation of the patient's tissue.
For a long standing technology, a shorter description of the history of the technology (with appropriate references) could be used. For example, the CER for a new wound dressing would not need to trace the history of wound dressings all the way back to the Romans.
Clearly state if the clinical data used in the evaluation are for an equivalent device. Identify the equivalent device(s) and provide a justification of the equivalency, cross-referenced to the relevant non-clinical documentation that supports the claim. Not to be confused with substantial equivalence, MEDDEV 2.7.1 r3 equivalence (page 42) means, "1) Clinically: used for the same clinical condition or purpose, at the same site in the body, in similar population (including age, anatomy, physiology); have similar relevant critical performance according to expected clinical effect for specific intended use, 2) Technically: used under similar conditions of use; have similar specifications and properties e.g. tensile strength, viscosity, surface characteristics; be of similar design; use similar deployment methods (if relevant); have similar principles of operation, 3) Biologically: use same materials in contact with the same human tissues or body fluids."
State the Essential Requirements relevant to the device in question. In particular, describe any special design features that pose performance or safety concerns, e.g. the presence of medicinal, human or animal components. These features should be identified in the device risk management documentation as requiring assessment from a clinical perspective.
Sources of data
Outline the considerations used to choose the clinical databases for the evaluation. Usually we see publication sources such as PubMed or MAUDE, but rarely do we see a discussion of why these databases were chosen. Provide an outline of the search and retrieval process for scientific literature, including the actual key words and search strings, and cross-reference these to your standard procedure for searching and reporting the literature (you have one, of course.)
5. Summary of the clinical data and appraisal
Provide a tabulation of the clinical data used in the evaluation, categorized according to whether the data address the performance or the safety of the device in question. (Note: many individual data sets will address both safety and performance.) Within each category, order the data according to the importance of their contribution to establishing the safety and performance of the device and in relation to any specific claims about performance or safety. Additionally, provide a brief outline of the data appraisal methods used in the evaluation, including any weighting criteria, and a summary of the key results.
Include full citations for literature-based data and the titles and investigation codes (if relevant) of any clinical investigation reports. At CDG, we begin an independent Excel spreadsheet as soon as we start reviewing abstracts. The spreadsheet lists the complete title and citation for the abstract, and a justification if the full article is not reviewed further. The spreadsheet is provided as an attachment to the CER.
Cross-reference the entry for each item of data to its location in the manufacturer’s technical documentation.
6. Data analysis
6.1 Performance
Provide a description of the how the literature analysis, or analysis of sponsor-acquired clinical data, was used to assess performance.
Identify the datasets that are considered to be the most important in contributing to the demonstration of the overall performance of the device and, where useful, particular performance characteristics. Outline why they are considered to be “pivotal” datasets and how they demonstrate the performance of the device collectively (e.g. consistency of results, statistical significance, clinically significance of effects.)
6.2 Safety
Describe the total experience with the device, including numbers and characteristics of patients exposed to the device; and duration of follow-up of device recipients.
You might include the number of patient-days of exposure, if some patients are exposed or followed longer than others. You should explain if patients were lost to follow-up.
Provide a summary of device-related adverse effects, paying particular attention to serious adverse device effects.
Provide specific comment on whether the safety characteristics and intended purpose of the device require training of the end-user.
6.3 Product literature and instructions for use
State whether the manufacturer’s proposed product literature and Instructions for Use (IFU) are consistent with the clinical data. Does the literature cover all the hazards and other clinically relevant information that may impact on the use of the device?
Implanted or home use devices require special consideration, describe how patients will be educated on the care and use of their device and how they can obtain help or additional information, especially outside of business hours.
7 Conclusions
Outline clearly the conclusions reached about the safety and performance of the device from the evaluation, with respect to the intended use of the device. State whether the risks identified in the risk management documentation have been addressed by the clinical data.
For each proposed clinical indication state whether:
• the clinical evidence demonstrates conformity with relevant Essential Requirements;
• the performance and safety of the device as claimed have been established; and
• the risks associated with the use of the device are acceptable when weighed against the benefits to the patient
A word about risk assessments
The contents and process of assessing risk is poorly discussed in the MEDDEV 2.7.1 r3 guideline. Here we discuss the process as it is usually performed at CDG. We begin with examining the Instructions for Use (IFU) and the most current Risk Assessment Report (RAR) per
ISO 14971.
Do the clinical data reveal any new risks not already discussed? If yes, you may need to update the RAR and IFU.
[5. ISO 14971 2012]
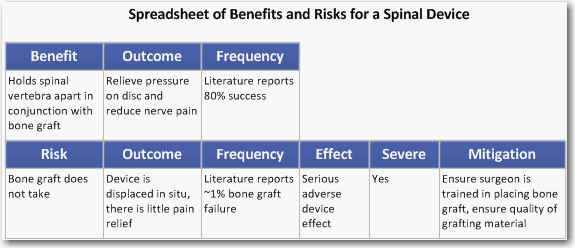
Preparing a spreadsheet listing the clinical benefits and risks is a good strategy for grasping their scope. Describe the nature of each benefit and risk in a column and the anticipated outcome in the next column. Indicate the anticipated frequency of occurrence of each benefit and risk in column three. In column four, identify type of adverse effect likely to occur. Next, indicate if the risk would result in a mild, moderate, or severe adverse effect; for example, a headache rarely meets the criteria of serious, yet it could be severe. Sometimes we indicate if the risk is associated with the procedure or the device itself. Finally, indicate how the risks have been mitigated. We are trying to gather together as much information as possible to understand the overall risk of the device.
Subjectively, we then compare the benefits and risks to determine if one outweighs the other. A number of factors may come into consideration. For example, let's say that two patients suffer from spondylolisthesis, a defect in the spine which causes vertebra to slip to one side of the body. Let's also assume that one patient is in her thirties and the other in her seventies. Knowing that it takes a year to recover from corrective surgery, spinal fusion may make sense for the younger patient for whom pain management over fifty years of remaining life is unreasonable. Spinal fusion may not make sense in the older patient, whose life expectancy is ten more years, and who does not want to spend 10% of their remaining lifetime recovering from surgery.
Notified Body Check List
After you've written the report you should check your work using the Notified Body checklist on page 35 of the MEDDEV guidance. Here you will find a thorough and detailed itemization of the points you should include in the report. If you meet all of the criteria in the checklist, a Notified Body will have little power to reject your evaluation based on format alone.
[6 MEDDEV 2.7.1 r3, p 35]
References
[1] MEDDEV 2.7.1 r3, Clinical Evaluation: A guide for manufacturers and Notified Bodies, 2009.
[2] Directive 2007/4/EC, Official Journal of the European Union, L 247/21.
[3] NBOG CL 2010-1 Checklist for audit of Notified Body's review of Clinical Data/Clinical Evaluation.
[4] MEDDEV 2.7.1 r3, p32.
[5] ISO 14971 Application of Risk Management to Medical Devices, 2012.
[6] MEDDEV 2.7.1 r3, p 35.
![]()
![]()
![]()
|
If you have received this message in error, or wish to unsubscribe from this list, please click here. If the link does not work in your system, please email us at cdginc@clinicaldevice.com. |
