The Fine Print:
Prepayment, including postage or courier fee, is required. All purchases are final. Please ask questions before you buy.
Copyrights and intellectual property rights are retained by Clinical Device Group. You may view a CD as often as you like, share it with your company colleagues, and retaine it for future training within your firm. CDs may not be copied, shared with individuals outside of the purchasing firm, or broadcast over private webcasts.
Certificates of Attendance and CEUs will be issued upon request and after return of Feedback Form.
The Goal Our goal is to build an online university where you can learn everything you need to know about medical device clinical and regulatory pre-approval issues.
Workshops The Workshop is our basic unit of instruction. Workshops are college-level training events consisting of lecture, application theory, and discussion. We focus on a specific topic related to clinical investigations, regulatory affairs, biological safety, and a range of other medical device pre-approval issues. Scroll down for course descriptions.
Workshops are offered on CD. The CDs are 3-4 hour events, some were recorded before live a professional audience, others were recorded in a studio to get better sound quality. You can order the CD below, view it as often as you like, share it with your colleagues, train new hires, and build your department library. You'll receive copies of the slides (as pdf files), relevant handouts, and supporting forms and documents. You'll also receive the correct Player for the recording, either an Adobe Connect, Webex, GoToMeeting, or Flash Player.
Instructor Workshops are taught by Nancy J Stark, PhD. Dr. Stark has over 25 years experience in device trials, serves as the US cochair to ISO 14155—Clinical Investigation of Medical Devices for Human Subjects, is a member of ISO TC 232—Learning Services for Non-formal Education and Training, was identified as one of 100 Notable People in Medical Devices by Medical Device & Diagnostics Industry magazine, and serves on the Editorial Advisory Board of MD&DI. You can learn more about her at Nancy J Stark.
Earning Certificates and CEUs The International Association of Continuing Education and Training (IACET) has specific learning standards to which CDG adheres. The learning standards include: 1) the learner must attend (listen to) at least 90% of the lecture, 2) the learner must demonstrate competence by way of a concluding quiz, and 3) the learner must give feedback about the course.
You are on your honor to listen to the course and study the materials. The quiz, provided on the CD, is created in Adobe Captivate and plays in a Flash player. It is automatically graded and provides an option to email the grades to CDG for recording. A Feedback form is provided in SurveyMonkey or Word. Following the IACET/ISO standards, one CEU is issued for ten contact hours.
Certificates are Issued in the Registered Learner's Name We understand that your administrative assistant might purchase the CD on behalf of the primary learner, but please give our registration manager the name and email address of the learner so the Certificate and CEUs are issued to the correct individual. One Certificate and CEUs are included with the purchase of each CD.
Additional Learners We are happy to offer Certificates and CEUs for additional learners at a modest price. Additional learners can be added at any time after the initial purchase, unless the material has undergone substantial changes to accurately reflect current regulations and practices.
System Requirements for CD Presentations 1. Computer.
2. Speakers or earphones.
3. Ability to play presentation. Either an Adobe Connect, Webex, GoToAssist, or Flash Player is provided on the CD, please check with your IT department if you have difficulty with installation—some companies have restrictions on new installations. If you still need assistance call CDG at 773-489-5706.
Fee:
CD: $550
Includes one learner.
Additional learners
i.e. certificates): $175
Agenda:
Lecture – 3 hours
Quiz – .5 hour
CEUs:
0.35 (IACET/ISO guidelines)
CD $550
The Food and Drug Agency (FDA) gains it authority to govern medical devices in interstate commerce from the Food, Drug, and Cosmetic Act. Drugs have been regulated since 1938, but medical devices weren't regulated until 1980. Devices are categorized as Class I, Class II, or Class III. Classification is based on whether or not there exists a substantially equivalent device (a predicate) already on the market in the United States. Congress did not design classification to be risk-based classification, a situation that is disconcerting to some activists.
Class I and Class II devices are cleared for commercialization by submitting to FDA a 510k (or "self-certifying" a 510k in some situations). The 510k presents the argument that the device is substantially equivalent to the named predicate. Class III devices, for which by definition there is no predicate, are approved for commercialization by submitting to FDA a PMA. The PMA establishes safety and efficacy de novo.
There are only two things that can go wrong with a medical device: it can be adulterated or it can be misbranded. Adulterated speaks to the issue of filth, contamination, or toxic leachables present in or on a medical device. Misbranding speaks to the issue of false or misleading statements, including statements made on websites or the juxtaposition of images. The surgical scalpel above is adulterated because it is not sterile (green bacteria) and mislabeled because the cutting edge has burrs (should be sharp). Adulteration or misbranding can lead to post-market adverse event reports to FDA's MAUDE database or even to product recalls.
This is an introductory workshop and designed for people new to medical devices. The objective is to learn the basics of FDA device regulations and how to determine if you need a clinical study in order to get FDA clearance.
Learn More ->
Fee:
CD: $550
Includes one learner.
Additional learners
i.e. certificates): $175
Agenda:
Lecture – 2 hours
Quiz – .5 hour
CEUs:
0.25 (IACET/ISO guidelines)
CD $550
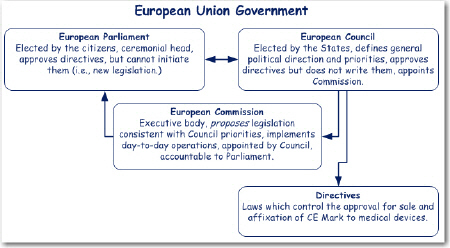
The European Union’s (EU) government, located in Brussels, consists of a Parliament, Council, and Commission. The Council sets the general direction and priorities of the Union. The Commission proposes directives consistent with the priorities set by the Council. The Parliament and Council jointly approve the directives, which are then issued as law by the Council.
When the EU sought to set up a review and approval system for medical devices, they wanted to take advantage of existing governmental institutions. The idea of using what was already there was called the New Approach. Under the New Approach, the European Council issued three primary directives (laws). These are the Active Implantable Medical Device Directive (AIMD) 1991-385-EEC, the Medical Device Directive (MDD) 1993-42-EEC, and the In Vitro Diagnostic Directive (IVD) 1998-79-EC.
The directives are actualized through three bodies unique to each Member State (country): the Regulatory Body (say, Health and Human Services) which implements the directives, the Standards Body which writes the ‘regulations’, and the Notified Body which inspects and enforces the directives and regulations. Because these bodies were unique to each country there was still considerable variability across Europe. So much so that the New Approach was challenged in 2009 and was nearly dissolved. The concept prevailed, however, but resulted in amendments to the directives that have had a big influence on device regulation in the EU.
This workshop is designed for Americans new to EU regulations. The objective of the workshop is to learn the basics of European regulations and how to determine if you need a clinical study in order to get market your device. Learn More ->
Fee:
CD: $550
Includes one learner.
Additional learners
i.e. certificates): $175
Agenda:
Lecture – 5 hours
Quiz – .5 hour
CEUs:
0.55 (IACET/ISO guidelines)
CD $550
The final and long-awaited version of ISO 14155 "Clinical investigation of medical devices for human subjects—Good clinical practice" was finally released in early 2011. The Part 1 and Part 2 standards from 2003 were combined and completely redesigned to make a single, comprehensive standard. Harmonized with the ICH-GCPs, the tag line "good clinical practice" is meant to signal the world that ISO 14155 is now the document to follow for international clinical studies of medical devices.
The approach is to discuss clinical research from a project management perspective and then identify the responsibilities of the sponsor and principal investigator in the last two sections. The contents of the protocol, case report forms, investigator's brochure, final report, and study documentation are specified in separate annexes.
Other major changes include the emphasis on risk management. A sponsor cannot be compliant with ISO 14155 without a fully functioning risk management system integrated into the clinical plan. Included in risk management is the requirement to record and report device deficiencies. If you are among the many who felt reporting all adverse events was too much, you may be frustrated to learn there is more reporting required now. The objective of the workshop is to learn the nuts and bolts of ISO 14155 (2011). Learn more->
Fee:
CD: $550
Includes one learner.
Additional learners
i.e. certificates): $175
Agenda:
Lecture – 5 hours
Quiz – .5 hour
CEUs:
0.55 (IACET/ISO guidelines)
CD $550
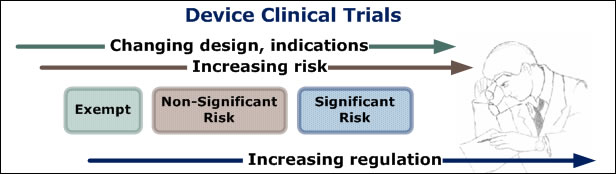
In the US, permission to conduct medical device clinical trials is obtained in one of two ways: 1) through a full Investigational Device Exemption (IDE) process for significant risk (SR) devices in which FDA reviews the protocol and approves the IDE application and study implementation, or 2) through an abbreviated IDE process for non-significant risk (NSR) devices in which the IRB acts as FDA's surrogate and reviews and approves the protocol for study implementation. Trials that are exempt from the regulations (Part 812) do not require FDA approval; although manufacturers that don't take advantage of this option are often surprised that it even exists.
The number of first-cycle approvals for IDE applications is under 40%. This means 60% are returned with questions. According to CDG's research, the majority of questions are regarding protocol or study design, the statistical analysis plan, and sample size calculations.
Delays in study implementation are costly because, at this point in development, management is thinking in terms of "lost opportunity" costs. Hence the pressure to obtain first-cycle approval is intense for study designers and regulatory professionals.
In this workshop you will learn how to prepare a significant risk IDE application for FDA, with the goal of achieving first-cycle approval. Learn More ->
This course will be based on new guidance documents from FDA, including: "Deciding When to Submit a 510k for a Change to an Existing Device"; "The 510k Program: Evaluating Substantial Equivalence in Premarket Notifications", "Medical Device Classification Product Codes", and more.
Fee:
CD: $550
Includes one learner.
Additional learners
i.e. certificates): $175
Agenda:
Lecture – 5 hours
Quiz – .5 hour
CEUs:
0.55 (IACET/ISO guidelines)
CD $550
A good study begins with team consensus on what you want to claim about your technology when the study is over. Writing clear statements about intended use and indications for use for efficacy or performance takes thought and debate. The difficulty is that you must commit today to what you will call a success tomorrow.
It helps to think in terms of the natural order of progression from want-to-have claims, to easy-to-get claims, to difficult-but-more-profitable claims. You’ll go for easy claims first if yours is a start-up firm; you may aim for the more difficult claims first if your firm is well-funded and experienced.
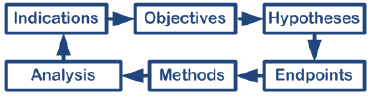
In this workshop, we’ll start by assessing the outcomes measures for product success or failure, and then look at the objectives for the study, the primary and secondary hypotheses, the primary and secondary endpoints, and how to measure them. These issues make up the creative heart of the investigation. Next we deal with the administrative text that gives the protocol standing. Finally we’ll take a layman’s look at data analysis. You will learn how to write a protocol that is consistent with FDA and international expectations. Learn more ->
Fee:
CD: $550
Includes one learner.
Additional learners
i.e. certificates): $175
Agenda:
Lecture – 5 hours
Quiz – .5 hour
CEUs:
0.55 (IACET/ISO guidelines)
CD $550
Think about what happens in a clinical trial. A manufacturer located in one state or country contracts with an investigator in another state or country to conduct a clinical trial that may last from a month to several years.
Like all human beings, investigators lose interest in trials, deviate from protocols to experiment with their own ideas, enroll subjects in competing studies, or generally misbehave—unless there is regular eyeball contact from the sponsor. Monitoring is the mechanism by which sponsors remain engaged with investigators. Monitors are the individuals charged with maintaining that eyeball contact.
What, exactly, do monitors do?
They prepare the site by assuring investigators and staff are trained, IRB and other approvals exist, and agreements and documents are in place (prestudy activities). The initiate the trial by observing first product applied to first subject and assuring compliance to the protocol during this process. They follow trial implementation for ongoing protocol and regulatory compliance and reporting of adverse events and trial deviations. Finally, they close out the trial by assuring the investigative site is inspection-ready. Their ongoing responsibility through all of this is reporting to the sponsor. Learn more ->
Fee:
CD: $550
Includes one learner.
Additional learners
i.e. certificates): $175
Agenda:
Lecture – 5 hours
Quiz – .5 hour
CEUs:
0.55 (IACET/ISO guidelines)
CD $550
Adverse event reporting is one of the more difficult aspects of conducting a medical device clinical trial. The US rules are different from drugs and from Europe. For example, in the States only adverse device effects are reportable (unless specifically required by FDA), while in Europe all adverse events are reportable, device related or not.
And in the States, clinical trials are regulated according to risk. Non-significant risk studies require only IRB approval and adverse events are only reported to the IRB, unless they are serious and unanticipated. But in Europe, all clinical trials are created equal and the same approval process is required no matter what the risk level of the study. Similarly all adverse events are reported to regulatory authorities.
Many device manufacturers also capture a type of "event" called a device deficiency alongside the adverse event collection. A device deficiency is pretty much what it sounds like: it is a failure or potential failure of the device, regardless of whether or not a subject was injured. Deficiencies include events such as investigator error and inaccurate labeling.
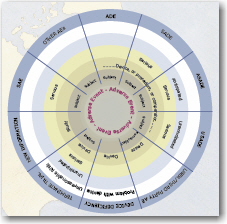
Yet another category of device adverse effects is effects that happen to third-parties such as investigators, caregivers, family members, or bystanders. Although extremely rare, the idea that adverse event and effect reporting is so far-reaching is surprising to people new to device studies. The Adverse Event Wheel, shown at left, is a useful look-up tool for the field and helps you keep the many categories and definitions straight.
The goal of this workshop is to learn a decision-making framework that will facilitate correct adverse event reporting, no matter where your trial is conducted. Learn more ->
Fee:
CD: $550
Includes one learner.
Additional learners
i.e. certificates): $175
Agenda:
Lecture – 3 hours
Quiz – 1 hour
CEUs:
0.4 (IACET/ISO guidelines)
CD $550
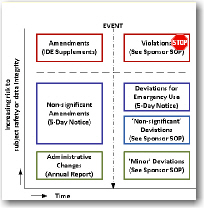
A deviation is an unplanned departure from the protocol or regulations. It is different from an adverse event in that injury to a subject or caregiver is not an issue and it is different from a device deficiency in that nothing about the device has failed.
IRBs call deviations "non-compliances" and they want to know about them. If an investigator has serious or repeated non-compliances an IRB may require them to take a training course or even prohibit them from being a primary investigator. Investigators are responsible for following the protocol and regulations. IRBs and sponsors are responsible for assuring they do so. And sponsors would be wise to assure investigator non-compliances are recorded and reported to IRBs.
An amendment is a planned and approved change to the protocol, including design changes to the investigational device or changes to its labeling. Only a perfectly designed study will not have amendments along the way.
Adverse events are closely related to deviations and amendments in that: 1) an event may be both non-compliant and adverse, 2) may be compliant and adverse, or 3) may be non-compliant and not adverse. In other words, there are overlaps.
The recording, reporting, and prior approval requirements for deviations and amendments are not always clear-cut, but your firm should have a clear-cut procedure on how to handle them. We have managed to organize deviations and amendments into a visual chart that will help you organize your thoughts.
In this workshop you will learn how to evaluate deviations, amendments, and non-compliances to the study and how to record and report them. Learn more ->
Fee:
CD: $550
Includes one learner.
Additional learners
i.e. certificates): $175
Agenda:
Lecture – 5 hours
Quiz – .5 hour
CEUs:
0.55 (IACET/ISO guidelines)
CD $550
Registry studies are observational studies in which the events which happen to subjects with a specific disease or condition are recorded without predefined treatment. It is a tricky concept to grasp: the protocol may specify data collection, and even diagnostic tests, but it does not specify a treatment plan.
This is in opposition to interventional studies in which subjects with a specific disease or condition are treated per an approved protocol and data are collected about the treatment. We say that registry studies are 'data driven' as opposed to interventional studies, which are 'protocol driven'.
One regulatory application for device registry studies is in the conduct of Section 522 post-market approval studies. But usually registry studies are purely at the option of the sponsor.
The workshop is based on " Registries for Evaluating Patient Outcomes: A User's Guide" developed by the Agency for Healthcare Research and Quality, which is the best ‘how to’ guide available for post-approval studies. You will learn when, why and how to implement a registry study and learn from some good device examples. Learn More ->
Fee:
CD: $550
Includes one learner.
Additional learners
i.e. certificates): $175
Agenda:
Lecture – 4 hours
Quiz – 1 hour
CEUs:
0.5 (IACET/ISO guidelines)
CD $550
The purpose of project management is to control the project and correct problems before they are uncorrectable. Without project management many trials evaporate into a cloud of no-enrollment and absence of data.
Projects are activities with beginnings, middles, and ends and close with work products, in contrast to programs which go on forever. We apply the six familiar steps of project management, 1) project definition, 2) project scheduling, 3) resource allocation, 4) cost estimation, 5) project maintenance, and 6) project close-out, to the process of implementing a clinical study as a way of understanding the enormity of the undertaking.
We begin by making a comprehensive task list for the project, where a task is a little activity with a beginning, middle, and end and which closes with a tangible work product. Once we make the task list they are arranged in chronological order as best as possible. Some tasks overlap, others cannot begin until others finish. Some tasks are repeated (several IRB approvals), others done only once (write a protocol). I like to group the tasks under the headings of pre-study, study, post-study, and data management tasks.
Next, you decide how long it will take it will take to perform each task. The more studies you perform the better you are at estimating. The more studies of a certain type of device, the better you are at estimating. But the timing is always an estimate and unexpected things will happen.
Next, the human and physical and financial resources required to implement each task is estimated. For example, how many monitoring visits will you need, how long will each monitoring visit last, and how many monitors must you hire?
Finally, we'll examine several methods of reporting on study progress to top management. Learn more ->
Fee:
CD: $550
Includes one learner.
Additional learners
i.e. certificates): $175
Agenda:
Lecture – 5 hours
Quiz – .5 hour
CEUs:
0.55 (IACET/ISO guidelines)
CD $550
A modest device study costs $1M and a major study $10M or more, big bucks for a start-up venture, not insignificant for even big firms. The total costs are divided between investigative sites, centers such as CROs or consultants, and internal costs such as your salary and benefits. Where does all that money go?
There are lots of mistakes we can make that cost us money. Here are twelve of them:
1) asking for unnecessary information,
2) incomplete or non-existent site task lists,
3) under-estimating the time required for a task,
4) under-estimating the cost of study procedures,
5) under-estimating overhead,
6) using under-trained monitors,
7) choosing centers by cost alone,
8) hiring a big firm for a small job,
9) hiring a small firm for a big job,
10) failing to monitor poorly performing sites,
11) not developing task, sub-task, and super-task lists to accommodate your colleague's personalities, and
12) failing to warn management that as much as 50% of the study budget will be spent before the first subject is enrolled.
In this workshop you'll learn how to develop a study budget, how to track expenses, and most importantly how to determine if your study is over or under budget.
Learn more->
Fee:
CD: $550
Includes one learner.
Additional learners
i.e. certificates): $175
Agenda:
Lecture – 5 hours
Quiz – .5 hour
CEUs:
0.55 (IACET/ISO guidelines)
CD $550
FDA has suggested device companies apply the quality management system regulations to clinical trials, the ICH-GCPs have always said clinical research functions should have procedures for conducting clinical trials, and now the ISO 14155 (2011) ‘Clinical investigation of medical devices in human subjects—good clinical practice’ standard requires procedures and a quality management system for clinical trials.
The key to a quality management system for a service function is to understand that your customers are the other functions within your firm. We'll look at how to identify the services (i.e. products) you provide to your customers and specify the quality of those work products. Usually 'quality' is measured in terms of compliance to regulations, guidances, and standards.
Quality management systems may be written broad and shallow (one procedure for one work product) or deep and narrow (one procedure for many work products, say one procedure for implementing an entire clinical trial.) We'll look at both styles so you can determine which is the best style for your firm. Then we'll consider a list of all the tasks necessary to include in the QMS.
We'll look at other sections of a QMS, such as vision and mission statements, and consider how they define the line between your job and the job function one department over. Very importantly, we'll look at a system or organizing clinical study documentation and what documentation should be maintained. Learn more ->
A complete Quality Management System for clinical research is available as a separate item for $3000. The CRQMS is described on the Book & Tools page.
Fee:
CD: $550
Includes one learner.
Additional learners
i.e. certificates): $175
Agenda:
Lecture – 4 hours
Quiz – .5 hour
CEUs:
0.45 (IACET/ISO guidelines)
CD $550 CD & Procedures $795
Clinical or literature evaluations are used in Europe to, 1) justify animal biological safety testing and preclinical studies, 2) justify the design of clinical investigations, and 3) apply to Notified Bodies for certification and CE Mark. They are reformatted, but are nevertheless an important part of IDE applications to FDA. And literature evaluations are contracted by the Agency for Healthcare Research and Quality (AHRQ) for technology assessments and comparative effectiveness reviews to assist the Center for Medicare and Medicaid in making reimbursement decisions. These evaluations play an increasingly critical role in the adoption and reimbursement success of medical technologies.
Becoming a skilled writer/critic of clinical evaluations takes effort, time, and training. This well-researched presentation takes you step-by-step through the process of writing a clinical evaluation by making it organized and understandable. The 100+ content slides and supporting handouts explain the logistics of clinical evaluations, how to define objective and key questions, how to identify key words, identify and retrieve abstracts, filter and screen abstracts to identify articles, assess and weight the literature, and write the evaluation to address the key questions originally posed. A strategy is proposed to allow each evaluation to build upon the previous ones. Finally, some average metrics are disclosed. Learn more ->
Fully-editable procedures, report templates, and protocols—conforming to MEDDEV 2.7.1 rev3—are available as an add-on products. Learn more ->
Fee:
CD: $550
Includes one learner.
Additional learners
i.e. certificates): $175
Agenda:
Lecture – 3 hours
Quiz – 1 hour
CEUs:
0.4 (IACET/ISO guidelines)
CD $550
This course is designed especially for professionals who are migrating from the drug industry to the medical device industry. It assumes a solid knowledge of regulatory affairs, and focuses on the differences between drug permissions and device permissions.
There are many kinds of permissions in the medical device world. Permissions to conduct clinical trials are called Investigational Device Exemptions (IDEs) and there are two kinds, full and abbreviated. Abbreviated IDEs do not require FDA approval (only IRB approval) and there is no list or registry of who conducts them or how many are conducted. Full IDEs require approval from both FDA and the local IRBs before a study can be implemented. Deciding whether you need a full or abbreviated clinical trial, or any clinical trial at all, is the first difficult question you will face.
Permissions to commercialize medical devices are called Premarket Notifications or 510(k)s and Premarket Applications or PMAs. 510(k)s are 'cleared' by FDA while PMAs are 'approved' by FDA.
Deciding whether you need a 510(k) or PMA is the second difficult question you will face.
You don't exactly have a choice, but sometimes the labelling (i.e. the claims and indications for use) can be manipulated to move the device from one category to another.
And which pathway you choose becomes a strategic business decision.
In this 3-hour workshop—designed specifically for pharmaceutical people or others new to device regulations—you will learn the differences between the permission processes and learn when to use which one, when.